Usecase2: In silico genotyping of reference genomes¶
This usecase describes how Brioche can be used to extract equivalent genotypes from any number of desired reference genomes to add them to sample sets for additional analyses.
This usecase assumes that the user is familiar with the basic structure of the Brioche results and what the anchored genotypes VCF file looks like but does not require a brioche run to be complete yet
Generate in silico genotypes by mapping marker sequences to a reference genome.
Requirements¶
Brioche targets file
Either:
The genotype data file you have is in one of the following three forms:
Raw genotypes in the format below (tab-separated).
REF/ALTare defined as ACGT/ACGT, samples are columns, markers are rows, genotypes are coded as0,1,2,NCas missing data, andNas null alleles.
Name REF ALT Sample1 Sample2 etc Marker1 T C 2 1 Marker2 T G 0 N Marker3 A C NC 1
A VCF file (any VCF-compatible format).
A DArTseq report (1-row format).
If you just want to generate insilico genotypes without samples, set the following variable (line ~69 of
Additional_functions/Run_insilico_add.sh) Performreferencegenotypingonly=”yes”
Reference genome FASTA
An updated
params.configfile ready to run Brioche
5. A list of full paths/filenames to reference genomes to in silico genotype (see example_files/genomes_insilico.txt) This needs to be in the
file format genomepath chromosomematching file and the genome NCBI refseq accession number (GCF_XYX or if none is available leave as “” as the value will be ignored by Brioche if not a valid REFseq code) ) separated by single spaces. The chromosomematching file can be provided for each reference genome so that a target chrom matching file can be used. If not
needed, provide a link to either a single file, or the example file (see `` example_files/Chromesome-chromosome_mappings.csv``) or a pathway to an empty file with the same headers in the example file
Installation of Nextflow, conda, git, and R
Runfile¶
To run insilico genotyping, a .sh script file is provided in Additional_functions/Run_insilico_add.sh Additional instructions are provided in the script but the main components to change are.
The Slurm
sbatchheader commands (e.g.,#SBATCH --job-name="example_insilicorunning")Four different variable settings:
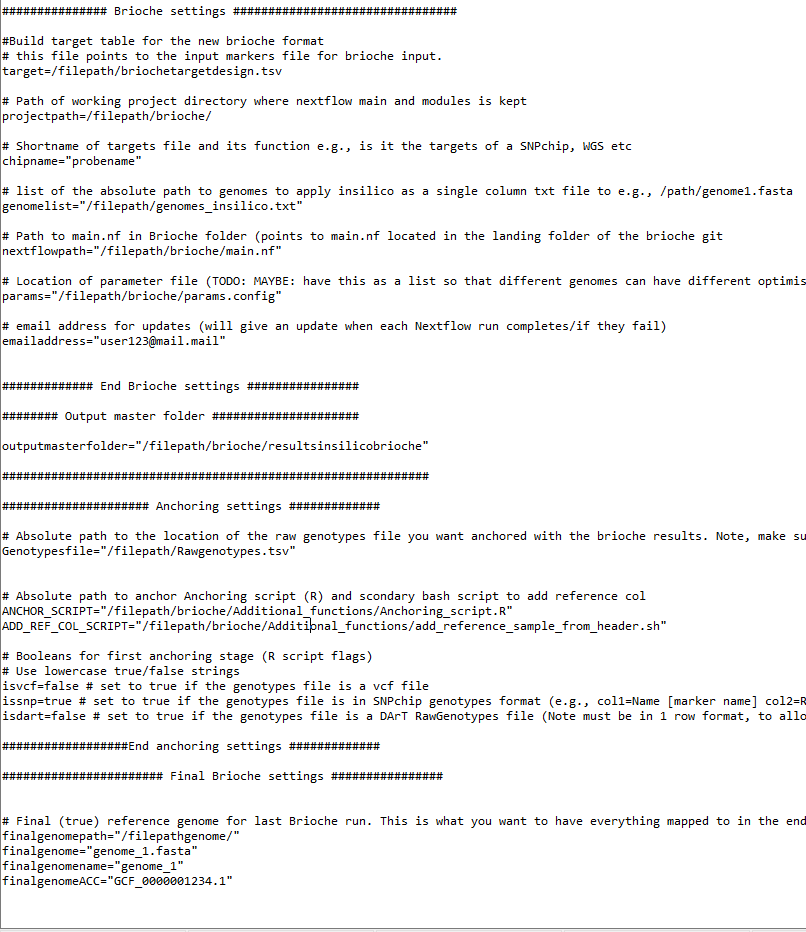
Brioche settings (lines 33-58, 7 settings). These settings require the user to update the paths to where Brioche was downloaded and where the targets file is located.
Output folder name (line 62, 1 setting). This setting describes where all in silico results should be saved.
Anchoring settings (lines 66-84, 7 settings). These settings point to the anchoring script and define the initial genotype format (e.g.,
isvcforisdartorissnp).Final Brioche settings (lines 86-97, 4 settings). These settings specify the final reference genome and what final VCF the genotypes will be anchored against.
Update the method for calling the required software (Nextflow, conda, git, and R) (lines 99-107). These are currently written as
module loadstatements.
Outputs¶
The results of insilico genotyping are output into three separate folders in the chosen output directory
intermediate_brioche/ where all Brioche runs are stored e.g., intermediate_brioche/genomename/brioche-results (one per reference genome)
anchoring/where all genomes are iteratively anchored into a single VCF
final/ where the final brioche run and final anchored results are stored.
For a detailed breakdown of 1) see Results folder structure
contains intermediate data and is not directly relevant for downstream analysis after Brioche
In the final folder the Brioche results and reports can be viewed but the primary result is in /anchored_results

In this folder there will be several output files of interest
‘Building_final_mapped_against_IWGSC_RefSeq_v2.1_refs.vcf’ A VCF of all markers containing the genotypes of the reference genomes (plus the two leading samples of the provided genotypes file)
‘Building_final_mapped_against_IWGSC_RefSeq_v2.1_seqs.vcf’ A VCF of all markers containing the genotypes of all samples from the genotypes file
‘Building_final_mapped_against_IWGSC_RefSeq_v2.1.vcf’ A VCF containing both the sequenced samples and the insilico genotyped references with markers which failed to map uniquely removed
‘Building_final_mapped_against_IWGSC_RefSeq_v2.1.vcf.gz’ A gzip of the above file
‘Building_final_mapped_against_IWGSC_RefSeq_v2.1.vcf_numericchroms.vcf’ A VCF with the name of chromosomes converted to numeric format
Chromosomes are converted to numeric format either through the user providing a chromosome matching file (see example_files/example_chrommatching_for_converting_vcf_chromnames_to_numeric.tsv) If no file is provided the chroms will be sorted through pattern matching names to look for numeric values If chromosomes lack numeric patterns, chromosomes will be renamed numerically from 1-n based on their order in the reference genome used
‘Building_final_mapped_against_IWGSC_RefSeq_v2.1.vcf_failedtomap.vcf’ A VCF of all markers which failed to map uniquely
‘unmapped_markers.tsv’ A one column tsv list of markers which failed to map uniquely
‘Building_final_mapped_against_IWGSC_RefSeq_v2.1.vcf_sorted.vcf’ A VCF copy of ‘Building_final_mapped_against_IWGSC_RefSeq_v2.1.vcf_numericchroms.vcf’ which has been sorted numerically by chromosome and position
Looking deeper into one of the markers we can see some of the novel results insilico genotyping can achieve. Below is a line containing only reference genome genotypes from an insilico run For this marker on chromosome 20 it can be seen that form the majority of genomes this marker has 2+ local duplications (the end value per genotype 0/0:0:2) This shows that the marker in question could represent a large variable duplication site and may be relevant if linked to genes of known function Alternatively, if the marker is in the middle of a region of non coding DNA this marker may be performing more poorly during sequencing because of the number of duplication sites)
‘20 744202465 Marker10 G A 100 PASS MAPSTATUS=Unique_mapping;PriorORIENT=plus;DUP=LocallyDuplicatedRegion;MAF=0;AN=25202;AC=0 GT:NU:DU 0/0:0:2 0/0:0:3 ./.:1:. 0/0:0:0 0/0:0:3 0/0:0:3 0/0:0:3 0/0:0:3 ./.:1:. 0/0:0:3 0/0:0:0’
Now the file ‘Building_final_mapped_against_IWGSC_RefSeq_v2.1.vcf_sorted.vcf’ can be taken and put into clustering analyses like PCAs to see where samples sit relative to known genomes etc.
Other usecases¶
If you are interested in other usecases see.
1. If you are interested in easy remapping of markers across any distinct reference genome and the reanchoring of genotypes to the new reference genome Usecase1: Remapping data across reference genomes
3. If you are interested in determining whether an existing marker dataset might be amplifying redundant regions under varied settings go to Usecase3: Testing redundancy/accuracy in marker datasets
4. If you are interested in the creation of custom marker datasets from existing analyses and testing the likely redundancy of newly designed markers against a wide range of reference genomes for a target species (similar process as 3.) go to Usecase3: Testing redundancy/accuracy in marker datasets
5. If you are interested in the mapping of multiple different datasets to a unified reference genome allowing for merging across shared loci and other downstream applications (e.g., imputations) Usecase4: Merge datasets
6. If you are interested in merging two VCF files mapped to the same reference genome and preserving the unique brioche chrUnk unmapped states go to Usecase5: Combining VCF files of different samples anchored against the same reference
otherwise, to return to the Introduction page go to Introduction
