Installation¶
Downloading brioche:¶
Brioche can be downloaded directly from the github page as shown below.
git clone https://github.com/plantinformatics/brioche.git
after downloading you can navigate to the brioche folder.
cd brioche
Here you will have a folder structure laid out like the following:
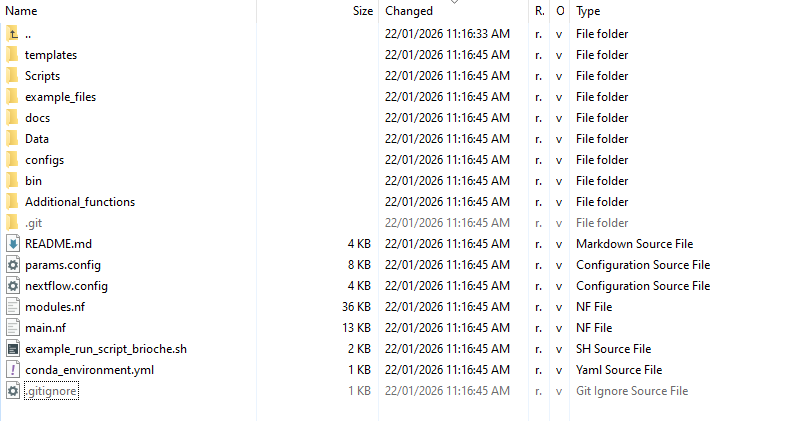
Software dependencies:¶
Brioche uses Nextflow as a workflow manager. Brioche also uses conda as a package manager and git for recording relevant metadata. Each of these are required to be installed either as modules on the HPC system or directly by the user.
e.g.,
module load Nextflow
or to download nextflow
wget -qO- https://get.nextflow.io | bash
The same applies for conda
module load Miniconda3
or to download conda we navigate to the desired location for storing conda (usually your home directory), download,unpack and initialise conda
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh -O /path/to/desired/directory/miniconda/miniconda.sh
bash /path/to/desired/directory/miniconda/miniconda.sh -b -u -p /path/to/desired/directory/miniconda/
/path/to/desired/directory/miniconda/bin/conda init bash
Finally, for git
module load git
or (asssuming Debian/Ubuntu Linux)
apt-get install git
Brioche is currently written to work within slurm HPC systems so will not run out of the box on other systems. If the user is interested in working with other HPC management systems individual .sh files will need to have their headers updated and the nextflow.config parameters updated manually.
During the first run of Brioche, the main conda environment will be built automatically from the conda_environment.yml file provided in the brioche directory. NOTE: this means that the launch partition of Brioche (e.g., example_run_script_brioche.sh) needs to have access to the internet so set the partition to one which can download.
Some additional software is required for extended functionality in Brioche. This can be found in the Additional_functions folder and will be installed as a conda environment using the file brioche-vcf.yaml This software environment is used to do additional insilico and other functionality.
This will install when running these scripts, or you can install it manually by running build_brioche-vcf_env.sh
File layout:¶
Brioche comes with a number of different files and folders but only a small number are important for the user to be aware of.
e.g., the following folders are usefull for the user
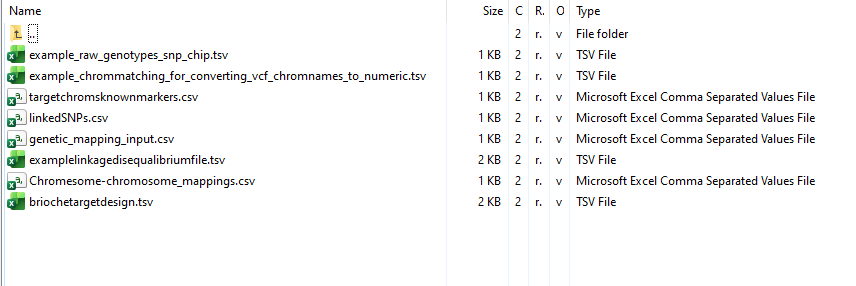
example_files/
example_files contains example formatted files for each optional and mandatory input and parameter file Brioche can use
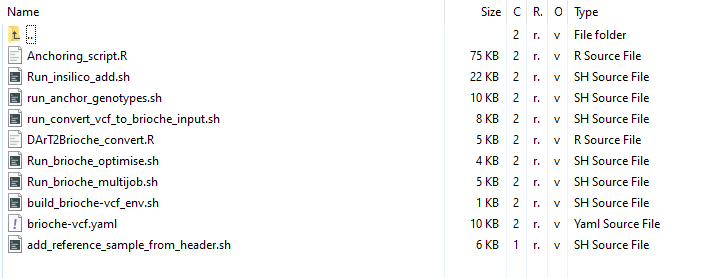
Additional_functions/
Additional_functions contains a collection of helpful bash and R scripts designed to expand the regular use of Brioche through 1. helping create input files from diverse datatypes (e.g.,run_convert_vcf_to_brioche_input.sh) and 2. Running specialised Brioche runs including testing the parameter space for marker uniqueness (e.g., Run_brioche_optimise.sh), or insilico adding reference genome genotypes (e.g., Run_insilico_add.sh)
Each of these scripts contains a short description of how to run in the file and what settings to change but additional details can be found in the relevant usecase described in this User guide.
The following files are useful for the user:
params.config
This file contains a list of all parameters that can be changed in Brioche allowing for greater control of a run
nextflow.config
This file contains additional details about resource allocation for the different types of runs Brioche can do. This can be edited to allocated great/less memory, CPUs etc per individual run if extremely large/small datasets and reference genomes are being used and standard allocations are not efficient.
example_run_script_brioche.sh
This file contains a simple run script for how to run Brioche. It can be opened, the slurm settings updated and then launched with sbatch commands.
Introduction:¶
To return to the Introduction page go to Introduction
Run setup:¶
For setting up a run go to Setting up a run
